
Non-Linear Function Approximation for Predicting Binding Affinity of PPAR-Targeting Antidiabetic Compounds from Molecular Descriptors
Article Sidebar
Main Article Content
 https://orcid.org/0000-0003-4478-6423
https://orcid.org/0000-0003-4478-6423
Abstract
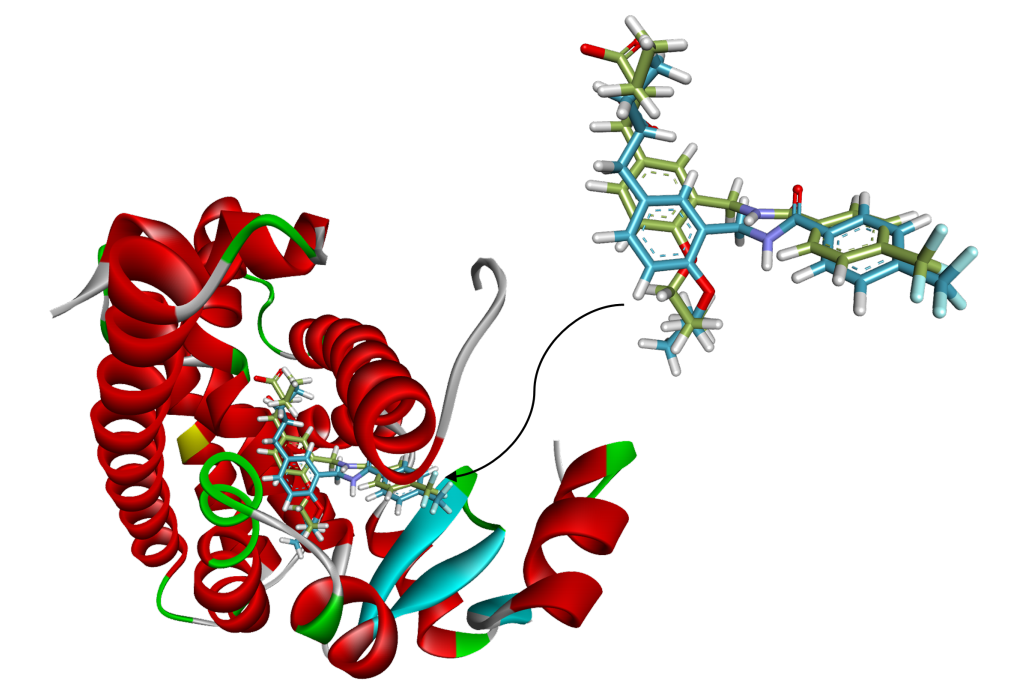
Diabetes mellitus remains a major global health challenge, necessitating the development of more effective therapeutic agents. The PPAR family plays a crucial role in regulating glucose and lipid metabolism, making it an important target for antidiabetic drug discovery. However, the identification of potent PPAR-targeting compounds is often limited by the high cost and time-consuming nature of experimental approaches. This study aims to develop a non-linear function approximation model to predict docking-derived binding affinity of antidiabetic compounds targeting PPAR using 2D molecular descriptors. A dataset of 3,764 small molecules with IC50 values was curated from the ChEMBL database, followed by data preprocessing to remove duplicates and incomplete entries. Molecular docking simulations were performed using AutoDock Vina to obtain binding affinity scores (kcal/mol), which were used as the target variable. Subsequently, 2D molecular descriptors were calculated from SMILES representations to capture key structural and physicochemical properties of the compounds. These descriptors were used as input features for a Multi-Layer Perceptron (MLP) regression model to approximate the complex non-linear relationship between molecular structure and binding affinity. The model achieved R² values of 0.853 for the training set and 0.632 for the test set, indicating moderate predictive performance and acceptable generalizability. Overall, this approach demonstrates the potential of machine learning as a cost-effective and scalable tool to support early-stage discovery of antidiabetic compounds targeting the PPAR family.
Article Details

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
References
[1] Ahmed I, El Turk S, Al Ghaferi A, Samad YA, Butt H. Nanocomposite Hydrogel-Based Optical Fiber Probe for Continu-ous Glucose Sensing. Small Science. 2024 feb;4(2). doi:10.1002/smsc.202300189.
[2] Ebere Ezinwanne Ilodibe, CU Nwankwo. Nurses’ competencies and resource availability in the care of diabetes mel- litus patient attending primary health care Centres in Anambra State. GSC Advanced Research and Reviews. 2023 feb;14(2):007-21. doi:10.30574/gscarr.2023.14.2.0038.
[3] Sahoo BR, Mohapatra G, Mohapatra J, Mohapatra N. Assessment of prevalence and pattern of comorbidities in hospitalized patients with uncontrolled hyperglycemia in Western Odisha. Multidisciplinary Science Journal. 2024 jul;6(3):2024015. doi:10.31893/MULTISCIENCE.2024015.
[4] IDF 11th. IDF Diabetes Atlas 11th Edition. In IDF Diabetes Atlas. vol. 11th editi. Brussels, Belgium: International Diabetes Federation; 2025.
[5] Haw JS, Galaviz KI, Straus AN, Kowalski AJ, Magee MJ, Weber MB, et al. Long-term sustainability of diabetes prevention approaches: A systematic review and meta-analysis of randomized clinical trials. JAMA Internal Medicine. 2017;177(12):1808-17. doi:10.1001/jamainternmed.2017.6040.
[6] Hassan FU, Nadeem A, Li Z, Javed M, Liu Q, Azhar J, et al. Role of peroxisome proliferator-activated receptors (Ppars) in energy homeostasis of dairy animals: Exploiting their modulation through nutrigenomic interventions. International Journal of Molecular Sciences. 2021 nov;22(22):12463. doi:10.3390/ijms222212463.
[7] Setia M, Meena K, Madaan A, Srikanth N, Dhiman KS, Sastry J. In vitro Studies on Antidiabetic Potential of New Dosage Forms of AYUSH 82. Journal of Drug Research in Ayurvedic Sciences. 2017;2(1):1-9. doi:10.5005/jp-journals-10059-0001.
[8] Laganà AS, Vitale SG, Nigro A, Sofo V, Salmeri FM, Rossetti P, et al. Pleiotropic actions of peroxisome proliferator- activated receptors (PPARs) in dysregulated metabolic homeostasis, inflammation and cancer: Current evidence and future perspectives. International Journal of Molecular Sciences. 2016;17(7):999. doi:10.3390/ijms17070999.
[9] Zhao Y, Gao P, Sun F, Li Q, Chen J, Yu H, et al. Sodium Intake Regulates Glucose Home- ostasis through the PPARδ /Adiponectin-Mediated SGLT2 Pathway. Cell Metabolism. 2016;23(4):699-711. doi:10.1016/j.cmet.2016.02.019.
[10] Peng L, Yang H, Ye Y, Ma Z, Kuhn C, Rahmeh M, et al. Role of peroxisome proliferator-activated receptors (Ppars) in trophoblast functions. International Journal of Molecular Sciences. 2021;22(1):1-13. doi:10.3390/ijms22010433.
[11] Przybycien´ P, Ga¸sior-Perczak D, Placha W. Cannabinoids and PPAR Ligands: The Future in Treatment of Polycystic Ovary Syndrome Women with Obesity and Reduced Fertility. Cells. 2022;11(16):2569. doi:10.3390/cells11162569. [12] Holm LJ, Mønsted MØ, Haupt-Jorgensen M, Buschard K. PPARs and the development of type 1 diabetes. PPAR Research. 2020;2020:1-11. doi:10.1155/2020/6198628.
[13] Chen M, Lin W, Ye R, Yi J, Zhao Z. PPARβ /δ Agonist Alleviates Diabetic Osteoporosis via Regulating M1/M2 Macrophage Polarization. Frontiers in Cell and Developmental Biology. 2021;9. doi:10.3389/fcell.2021.753194.
[14] Singh S, Kumar R, Payra S, Singh SK. Artificial Intelligence and Machine Learning in Pharmacological Research: Bridging the Gap Between Data and Drug Discovery. Cureus. 2023. doi:10.7759/cureus.44359.
[15] Mariam Z, Niazi SK, Magoola M. Optimizing Lead Compounds: The Role of Artificial Intelligence in Drug Discovery. Preprints. 2024:1-9. doi:10.20944/preprints202404.0055.v1.
[16] Unogwu OJ, Ike ME, Joktan OO. Employing Artificial Intelligence Methods in Drug Development: A New Era in Medicine. Mesopotamian Journal of Artificial Intelligence in Healthcare. 2023;2023:52-6. doi:10.58496/MJAIH/2023/010.
[17] Vora LK, Gholap AD, Jetha K, Thakur RRS, Solanki HK, Chavda VP. Artificial Intelligence in Pharmaceutical Technology and Drug Delivery Design. Pharmaceutics. 2023;15(7):1916. doi:10.3390/pharmaceutics15071916.
[18] Meli R, Morris GM, Biggin PC. Scoring Functions for Protein-Ligand Binding Affinity Prediction Using Structure-based Deep Learning: A Review. Frontiers in Bioinformatics. 2022;2. doi:10.3389/fbinf.2022.885983.
[19] Shen C, Ding J, Wang Z, Cao D, Ding X, Hou T. From machine learning to deep learning: Advances in scoring functions for protein–ligand docking. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2020;10(1):e1429. doi:10.1002/wcms.1429.
[20] Lee J, Yoon H, Lee YJ, Kim TY, Bahn G, Kim YH, et al. Drug–Target Interaction Deep Learning-Based Model Identi- fies the Flavonoid Troxerutin as a Candidate TRPV1 Antagonist. Applied Sciences (Switzerland). 2023;13(9):5617. doi:10.3390/app13095617.
[21] Shimizu Y, Ohta M, Ishida S, Terayama K, Osawa M, Honma T, et al. AI-driven molecular generation of not- patented pharmaceutical compounds using world open patent data. Journal of Cheminformatics. 2023;15(1). doi:10.1186/s13321-023-00791-z.
[22] Ridho M, Bustamam A, Adnan R. Reconstruction of the Phi-2 Method for Question-Answering Related to Di- abetes Disease Using the MedAlpaca Dataset. Jambura Journal of Biomathematics (JJBM). 2025;6(3):183-7. doi:10.37905/jjbm.v6i3.30506.
[23] Adekunle TA, Ogundoyin IK, Akanbi CO. Machine Learning Model for Predicting the Temporal Lassa Fever Confirmed Cases in Nigeria. Jambura Journal of Biomathematics (JJBM). 2025;6(3):166-72. doi:10.37905/jjbm.v6i3.33831.
[24] Jiang X, Lu L, Li J, Jiang J, Zhang J, Zhou S, et al. Synthetically Feasible De Novo Molecular Design of Leads Based on a Reinforcement Learning Model: AI-Assisted Discovery of an Anti-IBD Lead Targeting CXCR4. Journal of Medicinal Chemistry. 2024;67(12):10057-75. doi:10.1021/acs.jmedchem.4c00184.
[25] Rafeeq MM, Sain ZM, Alturki NA, Alzamami A, Asiri SA, Mashraqi MM, et al. Computational Screening of Natural Compounds for the Discovery of Potential Aromatase Inhibitors: A Promising Therapy for Estrogen-Dependent Breast Cancer. Journal of Pharmaceutical Research International. 2021:72-8. doi:10.9734/jpri/2021/v33i32a31717.
[26] Stjernschantz E, Oostenbrink C. Improved ligand-protein binding affinity predictions using multiple binding modes. Biophysical Journal. 2010;98(11):2682-91. doi:10.1016/j.bpj.2010.02.034.
[27] Erdas-Cicek O, Atac AO, Gurkan-Alp AS, Buyukbingol E, Alpaslan FN. Three-Dimensional Analysis of Binding Sites for Predicting Binding Affinities in Drug Design. Journal of Chemical Information and Modeling. 2019;59(11):4654-62. doi:10.1021/acs.jcim.9b00206.
[28] Jiménez-Luna J, Pérez-Benito L, Martínez-Rosell G, Sciabola S, Torella R, Tresadern G, et al. DeltaDelta neural networks for lead optimization of small molecule potency. Chemical Science. 2019;10(47):10911-8. doi:10.1039/c9sc04606b.
[29] Lecun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521(7553):436-44. doi:10.1038/nature14539.
[30] Chen H, Engkvist O, Wang Y, Olivecrona M, Blaschke T. The rise of deep learning in drug discovery. Drug Discovery Today. 2018;23(6):1241-50. doi:10.1016/j.drudis.2018.01.039.
[31] Lenselink EB, Ten Dijke N, Bongers B, Papadatos G, Van Vlijmen HWT, Kowalczyk W, et al. Beyond the hype: deep neu- ral networks outperform established methods using a ChEMBL bioactivity benchmark set. Journal of Cheminformatics. 2017;9(1):45. doi:10.1186/s13321-017-0232-0.
[32] Wieder O, Kohlbacher S, Kuenemann M, Garon A, Ducrot P, Seidel T, et al. A compact review of molec- ular property prediction with graph neural networks. Drug Discovery Today: Technologies. 2020;37:1-12. doi:10.1016/j.ddtec.2020.11.009.
[33] Schneider G, Clark DE. Automated De Novo Drug Design: Are We Nearly There Yet? Angewandte Chemie - Interna- tional Edition. 2019;58(32):10792-803. doi:10.1002/anie.201814681.
[34] Jiang D, Wu Z, Hsieh CY, Chen G, Liao B, Wang Z, et al. Could graph neural networks learn better molecular representa- tion for drug discovery? A comparison study of descriptor-based and graph-based models. Journal of Cheminformatics. 2021 feb;13(1):12. doi:10.1186/s13321-020-00479-8.
[35] Nair V, Hinton GE. Rectified linear units improve Restricted Boltzmann machines. In: ICML 2010 - Proceedings, 27th International Conference on Machine Learning; 2010. p. 807-14.
[36] Kingma DP, Ba JL. Adam: A method for stochastic optimization. 3rd International Conference on Learning Represen- tations, ICLR 2015 - Conference Track Proceedings. 2015.
[37] Stokes JM, Yang K, Swanson K, Jin W, Cubillos-Ruiz A, Donghia NM, et al. A Deep Learning Approach to Antibiotic Discovery. Cell. 2020;180(4):688-702.e13. doi:10.1016/j.cell.2020.01.021.
[38] Landrum G. RDKit: Open-source cheminformatics. RDKit. 2013.
[39] O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An Open chemical toolbox. Journal of Cheminformatics. 2011;3(10):33. doi:10.1186/1758-2946-3-33.
[40] Abadi M, Barham P, Chen J, Chen Z, Davis A, Dean J, et al. TensorFlow: A system for large-scale machine learning. Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation, OSDI 2016. 2016:265-83.
[41] Buitinck L, Louppe G, Blondel M, Pedregosa F, Mueller A, Grisel O, et al. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research. 2011;12(85):2825-30.
[42] McKinney W. Data Structures for Statistical Computing in Python. In: Proceedings of the 9th Python in Science Conference; 2010. p. 56-61. doi:10.25080/majora-92bf1922-00a.
[43] Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry. 2010;31(2):455-61. doi:10.1002/jcc.21334.
[44] Hunter JD. Matplotlib: A 2D graphics environment. Computing in Science and Engineering. 2007;9(3):90-5. doi:10.1109/MCSE.2007.55.
[45] Morris GM, Ruth H, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry. 2009;30(16):2785-91. doi:10.1002/jcc.21256.
[46] Oyama T, Takiguchi K, Miyachi H. Crystal structures of the ligand-binding domain of human peroxisome proliferator-activated receptor δ in complexes with phenylpropanoic acid derivatives and a pyridine carboxylic acid derivative. Acta Crystallographica Section F: Structural Biology Communications. 2022 feb;78(2):81-7. doi:10.1107/S2053230X22000449.
[47] Tyagi P, Singh HM, Ghosh B, Biswas SK. Virtual screening for plant-based inhibitors targeting SARS-CoV papain-like protease. Journal of Biomolecular Structure and Dynamics. 2011;29(4):634-49. doi:10.1080/07391102.2011.10508572.
[48] Ain QU, Aleksandrova A, Roessler FD, Ballester PJ. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2015;5(6):405-24. doi:10.1002/wcms.1225.
[49] Wu Z, Ramsundar B, Feinberg EN, Gomes J, Geniesse C, Pappu AS, et al. MoleculeNet: A benchmark for molecular machine learning. Chemical Science. 2018;9(2):513-30. doi:10.1039/c7sc02664a.